![]() A hasnyálmirigyrák kezelésére korlátozott számú kemoterápiás szer áll rendelkezésre, és sajnos csak mérsékelt aktivitást mutat. Ugyanakkor, hasonlóan az egyéb rosszindulatú daganatokhoz, a hasnyálmirigy daganatok esetében is ésszerűnek és hasznosnak tűnik az egyes daganatminták molekuláris jellemzése, és a terápia annak megfelelő „személyre szabott” tervezése.
A hasnyálmirigyrák kezelésére korlátozott számú kemoterápiás szer áll rendelkezésre, és sajnos csak mérsékelt aktivitást mutat. Ugyanakkor, hasonlóan az egyéb rosszindulatú daganatokhoz, a hasnyálmirigy daganatok esetében is ésszerűnek és hasznosnak tűnik az egyes daganatminták molekuláris jellemzése, és a terápia annak megfelelő „személyre szabott” tervezése.
Egy nemrégi amerikai vizsgálat („Know Your Tumor, KYT”) azt bizonyította, hogy azok a betegek, akik hozzájutottak a részletes molekuláris analízishez és molekuláris terápiához, szignifikánsan hosszabb ideig éltek, mint azok, akik vagy nem jutottak hozzá a vizsgálathoz, vagy ha hozzá is jutottak, nem részesültek molekuláris célzott kezelésben. A célzott terápia alkalmazása az előrehaladott hasnyálmirigy daganatos betegek élettartamát mintegy egy évvel hosszabbította meg. Számos adat utal arra, hogy a hosszútávú gyógyszeres kezelésre szoruló hasnyálmirigy daganatos betegeknél érdemes mind a beteg örökölt genetikai állományának génhiba irányában történő vizsgálatát, mind a daganatminta molekuláris vizsgálatát elvégezni abban a reményben, hogy az egyéni eltérés feltárásával a beteg életkilátásait javító kezelésre nyílik lehetőség. Ma már mindkét eljárást nemzetközi ajánlások javasolják; a törekvés megvalósítását egyelőre világszerte elégtelen orvosi genetika kapacitás hiány és a molekuláris diagnosztikával történő lépéstartás nehézsége korlátozza, nehezíti. Nagy centrumokban azonban feltétlenül nyílik lehetőség a vizsgálatokra még Magyarországon is. Milyen vizsgálatokról van tehát szó?
1. Örökletes génmutációk vizsgálata
A hasnyálmirigyrákok mintegy 10-20 százalékában örökletes génhiba áll a betegség hátterében. Ilyen esetben sokszor a család többi tagjánál is előfordul(t) hasnyálmirigydaganat vagy valamilyen egyéb tipikus daganat (emlő-, petefészek, vagy más gyomor-bélrendszeri daganat), ugyanakkor ez nem kötelező érvényű. A hasnyálmirigyrákra (is) hajlamosító örökletes génhibák a következők lehetnek: BRCA1, BRCA2, ATM, PALB2, MLH1, MSH2, MSH6, PMS2, CDKN2A, és TP53: Li Fraumeni syndroma, STK11: Peutz-Jeghers syndroma és a családi halmozódást mutató hasnyálmirigy gyulladás, familiaris pancreatitis (géneltérések a PRSS1 vagy SPINK1 génekben). Az eltérések vizsgálatára orvosi genetikai tanácsadás, a géneltérés jelenlétének felmérése után kerülhet sor, amennyiben azt a beteg vagy egészséges egyén óhajtja igénybe venni. Az örökletes hajlamot „egyszerű” vérvizsgálattal vagy akár nyálminta vizsgálattal tudják igazolni. Ilyenkor több hetet igénybe vevő bonyolult laboratóriumi vizsgálatok alapján lehet megállapítani, hogy van-e olyan genetikai eltérés, mely hajlamosít a betegségre, vagy akár más daganatok megjelenésére. Magát a génvizsgálatot úgynevezett újgenerációs szekvenálással (NGS) végzik, és az esetleges talált hibát további molekuláris biológiai vizsgálattal erősítik meg. A felsorolt gének működési gyengeségének kimutatása (többnyire két különböző DNS károsodás/hiba javítás funkció csökkenése) a már megbetegedett egyének esetében egy-egy új kezelési eljárás sikerét vetíti előre. Az örökletes genetikai hajlam igazolása természetesen felveti a család egyéb tagjainak lehetséges érintettségét, további vizsgálatát is.
Örökletes génhibák és hasnyálmirigyrák kockázat
| Örökletes génhiba | Gyakoriság hasnyálmirigy rákos betegek körében | Gyakoriság hasnyálmirigy rákos betegek körében, ha a családban emlő-, petefészek- vagy hasnyálmirigy rák fiatal életkorban jelen volt | Hasnyálmirigyrák kockázata |
| BRCA2 | 1.4-17% | 16% | RR: 3.5-10x |
| BRCA1 | 0.35-1% | 2% | RR: 2.7-4.1x |
| PALB2 | 0.2-0.4% | 1% | RR: 6x |
| ATM | 0.5-2.3% | 2.6-3.4% | RR: 2x |
| CDKN2A | 1% | 2.5% |
RR: 20-46.6x Élethosszra vonatkozó kockázat: 17% |
| MLH1, MSH2, MSH6, PMS2, EPCAM (Lynch syndroma) | 0.5-1% |
RR: 9-11x Élethosszra vonatkozó kockázat: 4% |
|
| STK11 (Peutz-Jegherssyndrome) |
RR: 132x Élethosszra vonatkozó kockázat: 11-36% |
||
| PRSS1, CTFR, SPINK1 (Familiarispancreatitis) |
RR: 26-87x Élethosszra vonatkozó kockázat: 40-53% |
RR: relatív kockázat (az átlagnépességhez viszonyított kockázat)
Klein AP. Pancreatic cancer epidemiology: understanding the role of lifestyle and inherited risk factors. Nat Rev Gastroenterol Hepatol. 2021 Jul;18(7):493-502. doi: 10.1038/s41575-021-00457-x.
Tempero MA et al. Pancreatic Adenocarcinoma, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2021 Apr 1;19(4):439-457. doi: 10.6004/jnccn.2021.0017.
Daly MB et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic, Version 1.2020. J Natl Compr Canc Netw. 2020 Apr;18(4):380-391. doi: 10.6004/jnccn.2020.0017.
Choda A et al. Current Approaches to Pancreatic Cancer Screening. Am J Pathol 2019 Jan;189(1):22-35. doi: 10.1016/j.ajpath.2018.09.013
2. Daganatminta molekuláris genetikai vizsgálata
Mint korábban részleteztük, valamennyi rosszindulatú daganat kialakulásáért, fennmaradásáért genetikai változások felelnek. Ezek azonosítása az illető daganat sikeres kezelését segíti. A vizsgálat a daganatból vett mintán különféle molekuláris biológiai módszerekkel történik, többek között újgenerációs szekvenálással (NGS), vagy immunhisztokémiai meghatározásokkal. Leggyakrabban (85%) a KRAS molekula különféle mutációiról van szó. A KRAS molekula a sejtműködés középpontjában áll, számos életfolyamatra hatással van, és a hasnyálmirigyrákok többségénél felelős a daganat kialakulásáért, fennmaradásáért (tipikus „driver” mutáció ebben a betegségben). A daganat nagyfokú KRAS-függőségét megszüntető gátlószer fejlesztésére számos törekvés létezik, egyelőre a hasnyálmirigyrákban csak a ritkán előforduló KRASG12C mutáció esetében megoldott.
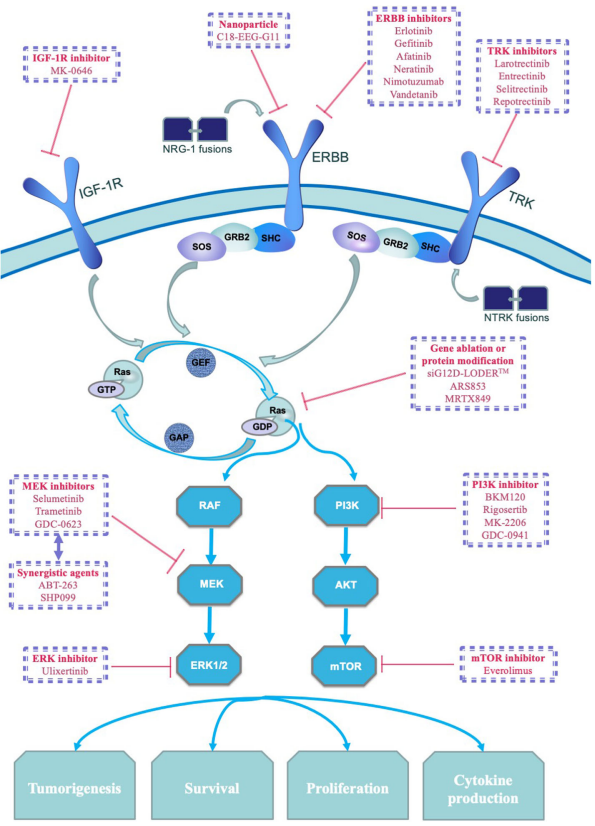 Különféle molekula-célpontok hasnyálmirigy ráksejtben, és a gyógyszeres kezelésben hasznosítható megfelelő hatóanyagok
Különféle molekula-célpontok hasnyálmirigy ráksejtben, és a gyógyszeres kezelésben hasznosítható megfelelő hatóanyagok
Forrás
A KRAS mutációja mellett ritkán előfordulhat más gén eltérése is, melynek gyógyszeres kezelése sikeres lehet. Legtöbbször a már az „Örökletes génmutációk vizsgálata” részben leírt gének örökletes, vagy csak a daganatban kimutatható (szomatikus) variánsáról van szó. A KRAS mutációktól mentes (KRAS vad típusú) daganatok esetében különösen gyakran fordul elő egyéb, támadható génhiba jelenléte.